

- 367.03 KB
- 26页


- 1、本文档共5页,可阅读全部内容。
- 2、本文档由网友投稿或网络整理,如有侵权请及时联系我们处理。
'分光光度法测铁含量一、实验目的⒈学习确定实验条件的方法,掌握邻二氮菲分光光度法测定微量铁的方法原理;⒉掌握721型分光光度计的使用方法,并了解此仪器的主要构造。二、实验原理⒈确定适宜的条件的原因:在可见光分光光度法的测定中,通常是将被测物与显色剂反应,使之生成有色物质,然后测其吸光度,进而求得被测物质的含量。因此,显色条件的完全程度和吸光度的测量条件都会影响到测量结果的准确性。为了使测定有较高的灵敏度和准确性,必须选择适宜的显色反应条件和仪器测量条件。通常所研究的显色反应条件有显色温度和时间,显色剂用量,显色液酸度,干扰物质的影响因素及消除等,但主要是测量波长和参比溶液的选择。对显色剂用量和测量波长的选择是该实验的内容。⒉如何确定适宜的条件:条件试验的一般步骤为改变其中一个因素,暂时固定其他因素,显色后测量相应溶液吸光度,通过吸光度与变化因素的曲线来确定适宜的条件。⒊本试验测定工业盐酸中铁含量的原理:根据朗伯-比耳定律:A=εbc。当入射光波长λ及光程b一定时,在一定浓度范围内,有色物质的吸光度A与该物质的浓度c成正比。只要绘出以吸光度A为纵坐标,浓度c为横坐标的标准曲线,测出试液的吸光度,就可以由标准曲线查得对应的浓度值,即工业盐酸中铁的含量。⒋邻二氮菲法的优点:用分光光度法测定试样中的微量铁,目前一般采用邻
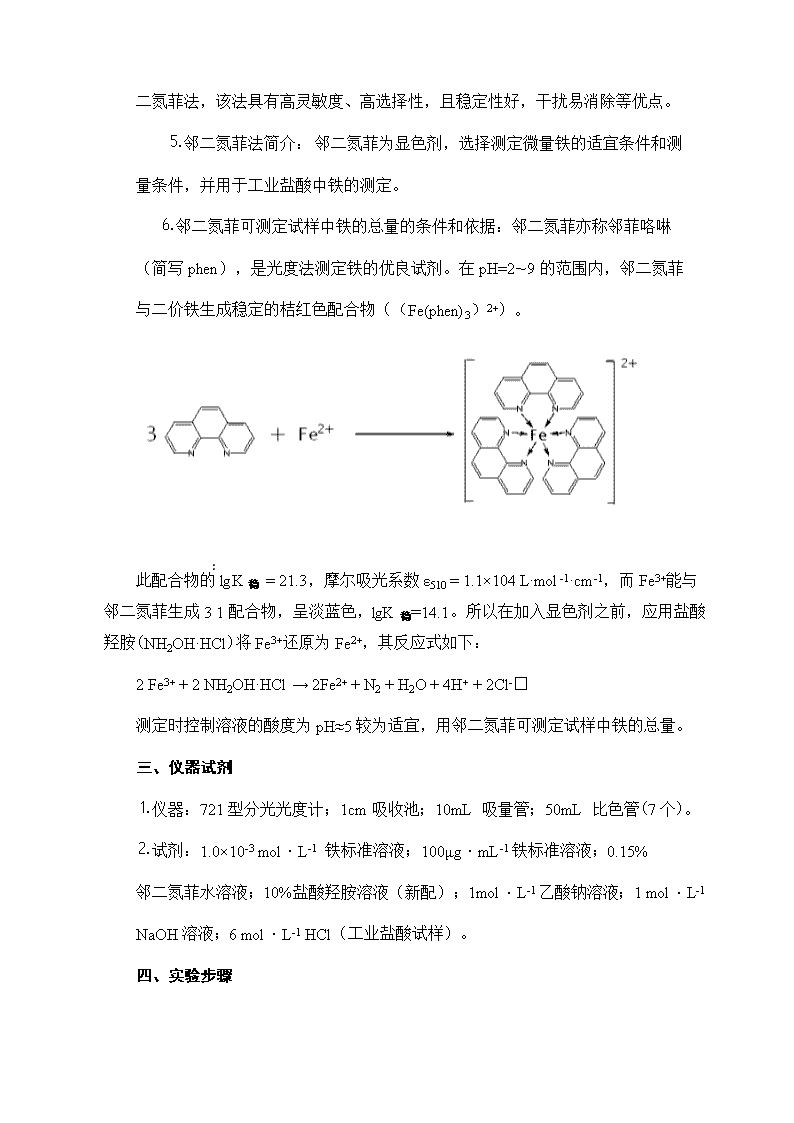
二氮菲法,该法具有高灵敏度、高选择性,且稳定性好,干扰易消除等优点。⒌邻二氮菲法简介: 邻二氮菲为显色剂,选择测定微量铁的适宜条件和测量条件,并用于工业盐酸中铁的测定。⒍邻二氮菲可测定试样中铁的总量的条件和依据:邻二氮菲亦称邻菲咯啉(简写phen),是光度法测定铁的优良试剂。在pH=2~9的范围内,邻二氮菲与二价铁生成稳定的桔红色配合物((Fe(phen)3)2+)。此配合物的lgK稳=21.3,摩尔吸光系数ε510=1.1×104L·mol-1·cm-1,而Fe3+能与邻二氮菲生成3∶1配合物,呈淡蓝色,lgK稳=14.1。所以在加入显色剂之前,应用盐酸羟胺(NH2OH·HCl)将Fe3+还原为Fe2+,其反应式如下:2Fe3+ +2NH2OH·HCl→2Fe2++N2+H2O+4H+ +2Cl-测定时控制溶液的酸度为pH≈5较为适宜,用邻二氮菲可测定试样中铁的总量。三、仪器试剂⒈仪器:721型分光光度计;1cm吸收池;10mL吸量管;50mL比色管(7个)。⒉试剂:1.0×10-3mol·L-1铁标准溶液;100μg·mL-1铁标准溶液;0.15%邻二氮菲水溶液;10%盐酸羟胺溶液(新配);1mol·L-1乙酸钠溶液;1mol·L-1NaOH溶液;6mol·L-1HCl(工业盐酸试样)。四、实验步骤
(一)准备工作打开仪器电源开关,预热,调解仪器。(二)测量工作(以通过空白溶液的透射光强度为I0,通过待测液的透射光强度为I,由仪器给出透射比T,再由T值算出吸光度A值)⒈吸收曲线的绘制和测量波长的选择用吸量管吸取2.00mL1.0×10-3mol·L-1铁标准溶液,注入50mL比色管中,加入1.00mL10%盐酸羟胺溶液,摇匀,加入2.00mL0.15%邻二氮菲溶液,5.0mLNaAc溶液,以水稀释至刻度。在光度计上用1cm比色皿,采用试剂溶液为参比溶液,在440~560nm间,每隔10nm测量一次吸光度(在最大吸收波长处,每隔2nm),以波长为横坐标,吸光度为纵坐标,绘制吸收曲线,选择测量的适宜波长。⒉显色剂条件的选择(显色剂用量)在6支比色管中,各加入2.00mL1.0×10-3mol·L-1铁标准溶液和1.00mL10%盐酸羟胺溶液,摇匀。分别加入0.10,0.50,1.00,2.00,3.00及4.00mL0.15%邻二氮菲溶液,5.0mLNaAc溶液,以水稀释至刻度,摇匀。在光度计上用1cm比色皿,采用试剂溶液为参比溶液,测吸光度。以邻二氮菲体积为横坐标,吸光度为纵坐标,绘制吸光度-试剂用量曲线,从而确定最佳显色剂用量。⒊工业盐酸中铁含量的测定⑴标准曲线的制作在6支50mL比色管中,分别加入0.00、0.20、0.40、0.60、0.80、1.00mL100μg/mL铁标准溶液,再加入1.00mL10%盐酸羟胺溶液,2.00mL0.15%邻二氮菲溶液和5.0mLNaAc溶液,以水稀释至刻度,摇匀。在512nm处,用1cm比色
皿,以试剂空白为参比,测吸光度A。⑵试样测定准确吸取适量工业盐酸三份,按标准曲线的操作步骤,测定其吸光度。五、实验数据记录与处理⒈标准曲线的制作波长(nm)440450460470480490492494496T(%)58.054.850.846.044.943.242.842.042.0吸光度A0.23660.26000.29000.33720.34780.36450.36860.37680.3768波长(nm)498.0500.0502.0504.0506.0508.0510.0512.0514.0T(%)41.841.641.040.041.140.039.839.239.5吸光度A0.37880.38090.38720.39790.38620.38720.40010.40670.4034波长(nm)516.0518.0520.0530.0540.0550.0560.0T(%)39.840.040.948.057.273.183.1吸光度A0.40010.39790.38820.31880.24260.13610.0804根据上面数据,作得标准曲线图如下:
由作图可知,最大吸收波长为512nm。⒉显色剂用量的测定邻二氮菲用量曲线:(λ=512nm)邻二氮菲的体积(mL)0.100.501.002.003.004.00透射比T(%)85.151.136.435.936.136.8吸光度A0.07010.29160.43890.4450.44250.4342据上面数据,作得标准曲线图如下: 由图可知,显色剂最佳用量为2.00mL0.15%邻二氮菲溶液。⒊工业盐酸中铁含量的测定⑴标准曲线的制作铁标液体积(mL)00.20.40.60.81.0铁浓度(μg/mL)00.40.81.21.62.0透射比T(%)10085.066.357.841.138.1吸光度A00.07060.17850.23810.38620.4191据上面数据,作得标准曲线图如下:
标准曲线方程为 y=0.2216*x-0.0061R2=0.98⑵试样测定(工业盐酸铁含量的测定)未知样标号12吸入盐酸的量(mL)1.01.5透射比T(%)82.275.1吸光度A0.08510.1244稀释后盐酸中铁的0.41160.5889含量(μg/mL)把y=0.0851,0.1244代入标准曲线方程y=0.2216*x-0.0061中,得到x=0.4116,0.5889,即稀释后工业盐酸中铁的含量为0.4116μg/mL,0.5889μg/mL。由1.0*Cx2/50=0.4116,得Cx1=20.58(μg/mL)由1.5Cx3/50=0.5889,得Cx2=19.63(μg/mL)则Cx=(Cx1+Cx2)/2=(20.58+19.63)/2=20.11(μg/mL)RSD=3.34%所以工业盐酸中铁的含量为20.11μg/mL。小结:通过实验数据可知,最适宜波长为λ=512nm;邻二氮菲(0.15%)的适
宜用量为2mL;最终由标准曲线得工业盐酸中铁的含量为:20.11μg/mL。六、实验注意事项⒈不能颠倒各种试剂的加入顺序。⒉读数据时要注意A和T所对应的数据。透射比与吸光度的关系为:A=log(I0/I)=log(1/T);测定条件指:测定波长和参比溶液的选择。⒊最佳波长选择好后不要再改变。⒋每次测定前要注意调满刻度。七、思考题⒈邻二氮菲分光光度法测定微量铁时为何要加入盐酸羟胺溶液?答:工业盐酸中含有Fe2+和Fe3+,其中Fe2+与邻二氮菲(phen)能生成稳定的桔红色配合物[Fe(phen)3]2+此配合物的lgK稳=21.3,摩尔吸光系数ε510=1.1×104L·mol-1·cm-1,而Fe3+能与邻二氮菲生成3∶1配合物,呈淡蓝色,lgK稳=14.1。所以在加入显色剂之前,应用盐酸羟胺(NH2OH·HCl)将Fe3+还原为Fe2+,然后,进行铁的总量的测定。2参比溶液的作用是什么?在本实验中可否用蒸馏水作参比?答:参比溶液的作用是扣除背景干扰,不能用蒸馏水作参比,因为蒸馏水成分与试液成分相差太远,只有参比和试液成分尽可能相近,测量的误差才会越小。3邻二氮菲与铁的显色反应,其主要条件有哪些?答:邻二氮菲与铁的显色反应,其主要条件有:酸度一般(PH=5~6)、温度、邻二氮菲的用量,显色时间等。八、实验总结通过本实验,学习了确定实验条件的方法,再次熟悉了可见分光光度法的测量原理和实验操作步骤,掌握了邻二氮菲分光光度法测定微量铁的方法原理以及掌握
721型分光光度计的使用方法。可见分光光度法基本要点:1.理解分子吸收光谱的产生及特征;2.理解光吸收基本定律和应用于紫外可见分光光度法的条件及其偏离因素;3.了解紫外-可见分光光度计的主要部件及其类型;4.理解紫外-可见分光光度法的显色反应条件和测量条件的选择;5.掌握紫外-可见分光光度法的定性分析和定量分析方法及其应用。·光学分析法光学分析法—利用辐射与物质间相互作用进行定性、定量的分析方法。光谱法光学光谱:原子吸收、紫外可见、荧光分析、原子发射等光学其它光谱:核磁共振、顺磁共振、X射线荧光等分析法非光谱法:折射法、偏振法、旋光法、园二向色散法、X射线衍射法等
·电磁波一.电磁波电磁波:实验证实,电磁波(电磁辐射)是一种以极高速度传播的光量子流。既具有粒子性,也具有波动性。1.波动性:其特征是每个光子具有一定的波长,可以用波的参数如波长(ë)、频率(í)、周期(T)、及振幅(A)等来描述。由于在真空中,所有电磁波均以同样的最大速度“C”传播,各种辐射在真空中有固定的波长:(1)但电磁波在任何介质中的传播速度都比在真空中小,通常用真空中的“l”值来标记各种不同的电磁波。波长单位:紫外可见区常用“nm”红外光区常用“㎛”微波区常用“cm”2.粒子性电磁辐射与物质之间能量的转移用粒子性来解释特征:辐射能是由一颗一颗不连续的粒子流传播的,这种粒子叫光量子,是量子化的(发射或被吸收)。光量子的能量:E=hn式中:h—plank常数,其值为6.626´10-34J·S光量子能量与波长的关系为:(2)例如:l为200nm的光,一个光量子的能量是:由于光量子能量小(10-19J),因此定义:1eV(电子伏)=1.6021´10-19J则上例中由(2)式可知:l®E¯,l¯®E即:随着l,辐射波动性变得较明显;
随着l¯,辐射的粒子性表现的较明显。二.电磁波电磁波谱:电磁辐射按波长顺序排列称为电磁波谱。紫外可见分光光度法:是根据物质分子对紫外及可见光谱区光辐射的吸收特征和吸收程度进行定性、定量的分析方法。·分子吸收光谱一.分子吸收光谱的产生(一)分子能级与电磁波谱分子中包含有原子和电子,分子、原子、电子都是运动着的物质,都具有能量,且都是量子化的。在一定的条件下,分子处于一定的运动状态,物质分子内部运动状态有三种形式:①电子运动:电子绕原子核作相对运动;②原子运动:分子中原子或原子团在其平衡位置上作相对振动;③分子转动:整个分子绕其重心作旋转运动。所以:分子的能量总和为E分子=Ee+Ev+Ej+⋯(E0+E平)(3)分子中各种不同运动状态都具有一定的能级。三种能级:电子能级E(基态E1与激发态E2)振动能级V=0,1,2,3⋯转动能级J=0,1,2,3⋯当分子吸收一个具有一定能量的光量子时,就有较低的能级基态能级E1跃迁到较高的能级及激发态能级E2,被吸收光子的能量必须与分子跃迁前后的能量差∆E恰好相等,否则不能被吸收。
图1双原子分子的三种能级跃迁示意图
对多数分子对应光子波长光谱∆E约为1~20eV1.25~0.06㎛紫外、可见区(电子)∆E约为0.5~1eV25~1.25㎛(中)红外区(振动)∆E约为10-4~0.05eV1.25cm~25㎛(远)红外区(转动)分子的能级跃迁是分子总能量的改变。当发生电子能级跃迁时,则同时伴随有振动能级和转动能级的改变,即“电子光谱”——均改变。因此,分子的“电子光谱”是由许多线光谱聚集在一起的带光谱组成的谱带,称为“带状光谱”。由于各种物质分子结构不同®对不同能量的光子有选择性吸收®吸收光子后产生的吸收光谱不同®利用物质的光谱进行物质分析的依据。二.紫外-可见吸收光谱与有机分子结构的关系(一)电子跃迁的类型许多有机化合物能吸收紫外-可见光辐射。有机化合物的紫外-可见吸收光谱主要是由分子中价电子的跃迁而产生的。分子中的价电子有:成键电子:s电子、p电子(轨道上能量低)未成键电子:n电子(轨道上能量较低)这三类电子都可能吸收一定的能量跃迁到能级较高的反键轨道上去,见图-3:键图2分子中价电子跃迁示意图1.s-s*跃迁
s-s*的能量差大®所需能量高®吸收峰在远紫外(l<150nm)饱和烃只有s、s*轨道,只能产生s-s*跃迁,例如:甲烷吸收峰在125nm;乙烷吸收峰在135nm(<150nm)(因空气中O2对<150nm辐射有吸收,定量分析时要求实验室有真空条件,要求一般难达到)2.p-p*跃迁p-p*能量差较小®所需能量较低®吸收峰紫外区(l200nm左右)不饱和烃类分子中有p电子,也有p*轨道,能产生p-p*跃迁:CH2=CH2,吸收峰165nm。(吸收系数e大,吸收强度大,属于强吸收)3.n-s*跃迁n-s*能量较低®收峰紫外区(l200nm左右)(与p-p*接近)含有杂原子团如:-OH,-NH2,-X,-S等的有机物分子中除能产生s-s*跃迁外,同时能产生n-s*跃迁,例如:三甲基胺(CH3)3N-的n-s*吸收峰在227nm,e约为900L/mol·cm,属于中强吸收。4.n-p*跃迁n-p*能量低®吸收峰在近紫外、可见区(l200~700nm)含有杂原子的不饱和基团,如-C=O,-CºN等,例如:丙酮:n-p*跃迁,lmax280nm左右(同时也可产生p-p*跃迁),属于弱吸收,e<500L/mol·cm.各种跃迁所需能量大小次序为:s-s*>n-s*³p-p*>n-p*紫外-可见吸收光谱法在有机化合物中应用主要以:p-p*、n-p*为基础。(二)吸收峰的长移和短移长移:吸收峰向长λ移动的现象,又称红移;短移:吸收峰向短λ移动的现象,又称紫移;增强效应:吸收强度增强的现象;减弱效应:吸收强度减弱的现象。(三)发色团和助色团p-p*、n-p*跃迁都需要有不饱和的官能团以提供p轨道,因此,轨道的存在是有机化合物在紫外-可见区产生吸收的前提条件。1.发色团:具有p轨道的不饱和官能团称为发色团。主要有:-C=O,-N=N-,-N=O,-CºC-等。
但是,只有简单双键的化合物生色作用很有限,其有时可能仍在远紫外区,若分子中具有单双键交替的“共轭大p键”(离域键)时,如:丁二稀CH2=CH—CH=CH2由于大p键中的电子在整个分子平面上运动,活动性增加,使p与p*间的能量差减小,使p-p*吸收峰长移,生色作用大大增强。2.助色团本身不“生色”,但能使生色团生色效应增强的官能团——称为助色团主要有:–OH、–NH2、–SH、–Cl、–Br等(具有未成键电子轨道n的饱和官能团)当这些基团单独存在时一般不吸收紫外-可见区的光辐射。但当它们与具有轨道的生色基团相结合时,将使生色团的吸收波长长移(红移),且使吸收强度增强。(助色团至少要有一对与生色团p电子作用的孤对电子)(四)溶剂效应(溶剂的极性对吸收带的影响)p-p*跃迁:溶剂的极性®长移三.吸收光谱吸收光谱:又称吸收曲线,是以波长(l)为横坐标、吸光度(A)为纵坐标所描绘的图形。特征:吸收峰曲线上比左右相邻处都高的一处;lmax吸收程度最大所对应的l(曲线最大峰处的l)谷曲线上比左右相邻处都低的一处;lmin最低谷所对应的l;肩峰介于峰与谷之间,形状像肩的弱吸收峰;末峰吸收在吸收光谱短波长端所呈现的强吸收而不呈峰形的部分。图3吸收曲线示意图
定性分析:吸收光谱的特征(形状和lmax)定量分析:一般选lmax测吸收程度(吸光度A)·光的吸收定律一.Lambert-Beer定律——光吸收基本定律“Lambert-Beer定律”是说明物质对单色光吸收的强弱与吸光物质的浓度(c)和液层厚度(b)间的关系的定律,是光吸收的基本定律,是紫外-可见光度法定量的基础。Lambert定律——吸收与液层厚度(b)间的关系Beer定律——吸收与物质的浓度(c)间的关系“Lambert-Beer定律”可简述如下:当一束平行的单色光通过含有均匀的吸光物质的吸收池(或气体、固体)时,光的一部分被溶液吸收,一部分透过溶液,一部分被吸收池表面反射;设:入射光强度为I0,吸收光强度为Ia,透过光强度为It,反射光强度为Ir,则它们之间的关系应为:I0=Ia+It+Ir(4)若吸收池的质量和厚度都相同,则Ir基本不变,在具体测定操作时Ir的影响可互相抵消(与吸光物质的c及b无关)上式可简化为:I0=Ia+It(5)(6)实验证明:当一束强度为I0的单色光通过浓度为c、液层厚度为b的溶液时,一部分光被溶液中的吸光物质吸收后透过光的强度为It,则它们之间的关系为:称为透光率,用T%表示。称为吸光度,用A表示则A=-lgT=K·b·c(7)此即Lambert-Beer定律数学表达式。L-B定律可表述为:当一束平行的单色光通过溶液时,溶液的吸光度(A)与溶液的浓度(C)和厚度(b)
的乘积成正比。它是分光光度法定量分析的依据。二.吸光度的加和性设某一波长(l)的辐射通过几个相同厚度的不同溶液c1,c2⋯⋯cn,其透射光强度分别为I1,I2⋯⋯In,根据吸光度定义:这一吸光体系的总吸光度为而各溶液的吸光度分别为:(8)吸光度的和为:(9)即几个(同厚度)溶液的吸光度等于各分层吸光度之和。如果溶液中同时含有n中吸光物质,只要各组分之间无相互作用(不因共存而改变本身的吸光特性),则:A=K1C1b1+K2C2b2+⋯⋯KnCnbn=A1+A2+⋯⋯+An(10)应用:①进行光度分析时,试剂或溶剂有吸收,则可由所测的总吸光度A中扣除,即以试剂或溶剂为空白的依据;②测定多组分混合物;③校正干扰。三.吸光系数Lambert-Beer定律中的比例系数“K”的物理意义是:吸光物质在单位浓度、单位厚度时的吸光度。一定条件(T、l及溶剂)下,K是物质的特征常数,是定性的依据。K在标准曲线上为斜率,是定量的依据。常有两种表示方法:1.摩尔吸光系数(e):当c用mol/L、b用cm为单位时,用摩尔吸光系数e表示,单位为L/mol·cmA=e·b·c(11)e与b及c无关。e一般不超过105数量级,通常:e>104为强吸收;e<102为弱吸收;102>e>104为中强吸收。吸收系数不可能直接用1mol/L浓度的吸光物质测量,一般是由较稀溶液的吸光系数换算得到。
2.吸光系数当c用g/L,b用cm为单位时,K用吸光系数a表示,单位为L/g·cmA=a·b·c(12)e与a之间的关系为:e=M·a(13)e——通常多用于研究分子结构a——多用于测定含量。四.引起偏离Lambert-Beer定律的因素根据L-B定律,A与c的关系应是一条通过原点的直线,称为“标准曲线”。但事实上往往容易发生偏离直线的现象而引起误差,尤其是在高浓度时。导致偏离L-B定律的因素主要有:1.吸收定律本身的局限性事实上,L-B定律是一个有限的定律,只有在稀溶液中才能成立。由于在高浓度时(通常C>0.01mol/L),吸收质点之间的平均距离缩小到一定程度,邻近质点彼此的电荷分布都会相互受到影响,此影响能改变它们对特定辐射的吸收能力,相互影响程度取决于C,因此,此现象可导致A与C线性关系发生偏差。此外,(n为折射率)只有当c£0.01mol/L(低浓度)时,n基本不变,才能用e代替e真。2.化学因素溶液中的溶质可因c的改变而有离解、缔合、配位以及与溶剂间的作用等原因而发生偏离L-B定律的现象。例:在水溶液中,Cr(Ⅵ)的两种离子存在如下平衡Cr2O42-+H2O⇌2CrO42-+2H+Cr2O42-、CrO42-有不同的A值,溶液的A值是二种离子的A之和。但由于随着浓度的改变(稀释)或改变溶液的pH值,[Cr2O42-]/[CrO42-]会发生变化,使C总与A总的关系偏离直线。消除方法:控制条件。3.仪器因素(非单色光的影响)L-B定律的重要前提是“单色光”,即只有一种波长的光;实际上,真正的单色光却难以得到。由于吸光物质对不同l的光的吸收能力不同(e不同),就导致对的偏离。“单色光”仅是一种理想情况,即使用棱镜或光栅等所得到的“单色光”实际上是有一定波长范围的光谱带,“单色光”
的纯度与狭逢宽度有关,狭缝越窄,他所包含的波长范围也越窄。4.其它光学因素(1)散射和反射:浑浊溶液由于散射光和反射光而偏离L-B(2)非平行光·分光光度计紫外-可见分光光度计是在紫外可见区可任意选择不同l的光测定吸光度的仪器。一.紫外-可见分光光度计的主要部件1.光源:提供入射光的装置;(1)钨灯或碘钨灯:发射光l范围宽,但紫外区很弱,通常取此l>350nm光为可见区光源(2)氢灯或氘灯:气体放电发光光源,发射150~400nm的连续光谱,用作紫外区同时配有:稳压电源(稳定I0);光强补偿装置;聚光镜等。2.单色器:将来自光源的光按波长的长短顺序分散为单色光并能随意调节所需波长光的一种装置。(1)色散元件——把混合光分散为单色光的元件是单色器的关键部分!)常用的元件有:棱镜——由玻璃或石英制成,它对不同l的光有不同的折射率,将复合光分开但:光谱疏密不均长l区密,短l区疏光栅——由抛光表面密刻许多平行条痕(槽)而制成,利用光的衍射作用和干扰作用使不同l的光有不同的方向,起到色散作用。(光栅色散后的光谱是均匀分布的)(2)狭缝——入口狭缝:限制杂散光进入出口狭缝:使色散后所需l的光通过(3)准直镜——以狭缝为焦点的聚光镜其作用为:将来自于入口狭缝的发散光变成单色光把来自于色散元件的平行光聚集于出口狭缝3.吸收池:装被测溶液用的无色、透明、耐腐蚀的池皿光学玻璃吸收池——只能用于可见区石英吸收池——可用于紫外及可见区。定量分析时:吸收池应配套(同种溶液测定∆A<0.5%)4.检测器:将接受到的光信号转变成电信号的元件。常用的有:(1)光电管一真空管内装有:一个丝状阳极——用镍制成
一个半圆筒状阴极——金属制成,凹面涂光敏物质。国产光电管:紫敏光电管:用锑、铯做阴极,适用范围200~625nm红敏光电管:用银、氧化铯作阴极,适用范围625~1000nm(2)光电倍增管:原理与光电管相似,结构上有差异。5.显示器:电表指针、数字显示、荧光屏显示等显示方式:A、T(%)、c等二.分光光度计的类型常见的可见及紫外-可见分光光度计:1.单波长、单光束分光光度计(721、722、752型等)一个单色器;一种波长的单色光;一束单色光。2.单波长双光束分光光度计从一个单色器获取一个波长的单色光用切光器分成二束强度相等的单色光实际测量到的吸光度A应为∆A(As-AR)(14)式中消去了I0,即消除了光源不稳定性引起的A值测量误差。3.双波长分光光度计二个单色器得到二个波长不同的单色光。两束波长不同的单色光(l1、l2)交替地通过同一试样溶液(同一吸收池)后照射到同一光电倍增管上,最后得到的是溶液对l1和l2两束光的吸光度差值∆A即Al1-Al2:图4双波长双光束分光光度计以双波长单光束方式工作时的光学系统图若用于测定浑浊样品或背景吸收较大的样品时,可提高测定的选择性,用AS表示非待测组分的吸光度(
背景吸收)则(15)(16)一般情况下:由于l1与l2相差很小,可视为相等(As一般不受l的影响,或影响甚微)∴As(1)=As(2)因此,通过吸收池后的光强度差为(17)该式表明:试样溶液中被测组分的浓度与两个波长l1和l2处的吸光度差∆A成比例,这是双波长法的定量依据。双波长分光光度计不仅可测定多组分混合试样、浑浊试样,而且还可测得导数光谱。·定性及定量分析方法一.定性分析选择合适的溶剂(非极性),使用有足够纯度单色光的分光光度计,在相同的条件下测定相近浓度的待测试样和标准品的溶液的吸收光谱,然后比较二者吸收光谱特征:吸收峰数目及位置、吸收谷及肩峰所在的位置(l)等;分子结构相同的化合物应有完全相同的吸收光谱。二.定量分析(一)单组分定量分析方法1.标准曲线法:配制一系列(5~10)个不同c的标准溶液,在适当l——通常为lmax下,以适当的空白溶液作参比,分别测定A,然后作A-c曲线同条件下测定试样溶液吸光度Ax,查找对应的cx。2.直接比较法:已知试样溶液基本组成,配制相同基体、相近浓度的标准溶液,分别测定吸光度A标、A样根据L-A定律:A标=K·b·c标A样=K·b·c样则(18)(二)多组分定量分析
混合组分的吸收光谱相互重叠的情况不同,测定方法也不相同,常见混合组分吸收光谱相干扰情况有以下三种:图5混合组分吸收光谱的三种相干情况示意图1.第一种情况:各种吸光物质吸收曲线不相互重叠或很少重叠,则可分别在l1及l2处测定a及b组分的c;2.第二种情况部分重叠:先在l1处测得ca,再在l2处测得混合组分的吸光度Aa+b,根据吸收定律加和性:即可求得cb。[应先求得ea(l2),与ea(l2),并使用相同b]3.第三种情况:两吸收曲线互相重叠,但服从L-B定律(1)解方程组法:若试样中需要测定两种组分,则选定两个波长l1及l2,测得试液的吸光度为A1和A2,则可解方程组求得组分a、b的浓度ca、cb:(在l1处)(在l2处)(19)如果混合物含有n个组分,可不经分离,在n个适当波长处进行n次测量,获得n个吸光度值,然后解n个联立方程以求得各组分的浓度。(2)等吸光度双波长(消去)法吸收光谱重叠的d、e两组分共存,现设法把一种组分(a)的吸光度消去。方法如下:
e图6二组分混合物吸收光谱用作图法选择l1、l2(双波长分光光度法)选取两个适当的波长l1和l2,使e1d=e2d,而尽可能大,则用这两个波长l1和l2测得混合物溶液吸光度之差∆A应只与ce成正比(而与cd无关),直接测得ce:因为所以若用1cm吸收池若需测定另一组分d时,也可用同样方法,选择另一个l1’和l2’,先消去的e的干扰,直接求cd·光度法显色反应条件和测量条件的选择一.影响显色反应的因素及反应条件的选择(一)显色反应的选择1.选择性好:干扰少或易排除;2.灵敏度高(S):尤其是对低含量组分,一般选择e:104~105L/mol·cm3.有色化合物稳定、组成恒定4.有色化合物与显色剂的颜色差别大(二)影响显色反应的因素及反应条件1.显色剂的用量
M+R⇌MR待测组分显色剂有色化合物在被测组分一定及其它实验条件不变的情况下,分别测得加入不同量显色剂测得A值,作A-cR曲线,常见以下二种情况:AcRcR图7吸光度与显色剂加入量的关系吸光度与显色剂加入量的关系(a)(b)在a与b之间任选一点严格控制CR因此,合适的cR通过实验确定。2.溶液的酸度(1)对金属离子存在状态的影响——防止水解,防止沉淀生成(2)对显色剂浓度的影响H2R⇌2H++R2-(3)对显色剂颜色的影响pKapKaH2R⇌H++HR-⇌2H++R2-6.912.4黄橙红适宜的pH通过实验确定:做A-pH曲线(其它条件并不变),从中找出A较大且基本不变的某pH范围。3.显色时间:各种显色反应得速度不同,反应完全所需时间不同;有些有色化合物在一定的时间内稳定。选择方法:作A-t(min)曲线,选择在A较大且稳定的时间内进行。4.显色温度:显色反应一般在室温下进行,但反应速度太慢或常温下不易进行的显色反应需要升温或降温。选择方法:作A-T(℃)曲线,选择在A较大的时间内进行。5.溶剂:实验确定——选择合适的溶剂(常为有机溶剂),提高反应的灵敏度及加快反应速度。二.分光光度法测量误差及实验条件的选择
(一)测量误差及A范围的选择任何一台分光光度计都有光度误差∆T%,但给定的一台分光光度计,∆T基本上是一常数,一般为±0.002~±0.01,但在不同T时同样的∆T对应的∆A则不同,所以引起的∆C/C(浓度的相对误差)就不同。由L-B定律得:(20)将此式微分得:浓度相对误差为:(21)设当∆T=±0.01时,不同T%时所对应的∆c/c可从相关表查得:当T%=36.8%即A=0.434时,∆c/c最小;当T%在15-65%之间即A在0.2~0.8范围内,∆c/c较小。实际测定时:可通过控制溶液的c及b使A在0.2~0.8范围内。(二)测量波长选择一般根据吸收光谱选择lmax测定——灵敏度高、A随波长变化小若有干扰,根据“吸收大,干扰小”原则选择l。如:3,3’-二氨基联苯(DAB)和Se形成配合物Se-DAB的最大吸收波长在340nm波长处,DAB也有很强的吸收,在这种情况下,分析波长应选用次大吸收波长420nm,否则测量误差较大。3.狭缝宽度理论上,定性分析采用最小的狭缝宽度,在定量分析中,为避免狭缝太小,出射光太弱而引起信噪比降低,可以将狭缝开大一点。通过测定A随狭缝宽度的变化规律,可选择出合适的狭缝宽度。狭缝宽度在某个范围内,A值恒定,狭缝宽度增大至一定程度时A减小,因此:合适的狭缝宽度是在吸光度不减小时的最大狭缝宽度。(三)空白溶液的选择空白溶液是用来调节工作零点即A=0,T%=100%的溶液,以消除溶液中其它基体组分以及吸收池和溶剂对入射光的反射和吸收所带来的误差。
根据情况不同,常用空白溶液有如下选择:1.溶剂空白:当溶液中只有待测组分在测定波长下有吸收,而其它组分无吸收时—用纯溶剂作空白;2.试剂空白:如果显色剂或其它试剂有吸收,而待测试样溶液无吸收—则用不加待测组分的其它试剂作空白;3.试样空白:如果试样基体有吸收,而显色剂或其它试剂无吸收—则用不加显色剂的试样溶液作空白;4.平行操作空白:用溶剂代替试样溶液,以与试样完全相同的分析步骤进行平行操作,用所得的溶液作空白。·紫外-可见分光光度法应用*酸碱指示剂离解常数的测定分光光度法可以测定酸碱离解常数,若为一元弱酸,在溶液中的离解反应为:HB⇌H++B-若测出[B-]和[HB],就可算出Ka。测定时,配制出三份不同pH的HB溶液,一份为强碱性溶液,另一份为强酸性溶液,分别在B-和HB的吸收峰波长处测定吸光度,由此计算出B-和HB的摩尔吸光系数。第三份为已知pH值的缓冲液,其pH值在pKa附近,在测得B-和HB的总吸光度后用双组分测定的方法算出B-和HB的浓度,即可计算出弱酸的离解常数。由pH=pKa可知,当[B-]和[HB]相等时:pKa=pH若以pH为横坐标,以某波长处测得的不同pH时的A为纵坐标作图,得一条S形曲线,该曲线的中点所对应的pH即为pK值。自测题1.什么是分光光度中的吸收曲线?制作吸收曲线的目的是什么?
2.什么是分光光度中的校准曲线?为什么一般不以透光度对浓度来制作校准曲线?3.试比较通常的分光光度法与双波长分光光度法的差别,并说明其理由。4.影响显色反应的条件有那些?怎样选择适宜的显色条件?5.浓度为1.02×10-4mol/L的酸碱指示剂HIn(Ka=1.42×10-5)水溶液,取2份,分别用等容的0.2mol/LNaOH与HCl0.2mol/L稀释后,用1.0cm吸收池在430nm处测得吸光度为1.51(碱性液),与0.032(酸性液)。计算此指示剂的纯水溶液浓度分别为2×10-5、4×10-5、12×10-5mol/L时在430nm处的吸光度。6.已知:Zn2+与螯合剂Q2-生成的配阴离子ZnQ22-在480nm有最大吸收。当螯合剂的浓度超过阳离子20倍以上时,可以认为Zn2+全部生成ZnQ22-。Zn2+或Q2-对480nm的光都不吸收。现有含Zn2+2.30×10-4mol/L与含Q2-9.60×10-3mol/L的溶液,用1.00cm吸收池于480nm处测得吸光度为0.690。同样条件下,只把螯合剂浓度改变为5.00×10-4mol/L,测得的吸光度为0.540。计算ZnQ22-的稳定常数。7.某指示剂HIn的离解常数是5.4×10-7,HIn的λmax是485nm,In-的λmax=625nm,今制成5.4×10-4mol/L溶液用1.00cm的吸收池在酸性与碱性下测得吸光度如下:吸光度(A)主要pH485nm625nm存在形式1.000.4540.176HIn13.000.0520.823In-问:(a)指示剂浓度不变而H+浓度为5.4×10-7mol/L时,在485nm与625nm处的吸光度将是多少?(b)指示剂浓度不变,改变溶液的pH值,在625nm处的吸光度是0.298,求溶液的H+浓度。'
您可能关注的文档
- 紫外分光光度法测定云南红豆杉枝叶中总黄酮的含量
- 分光光度法测定紫菀中总三萜类成分的含量论文
- 邻二氮菲分光光度法测定水中微量铁
- 磷钨钒酸分光光度法测定石煤中的钒
- 紫外分光光度法优选黄芩总黄酮的提取工艺研究
- 紫外分光光度法测定峰胶胶囊中总黄酮的含量
- 紫外分光光度法测定鹰嘴豆和豆芽中异黄酮含量的研究
- 酚试剂分光光度法测定甲醛的含量
- 水质氨氮的测定纳氏试剂分光光度法[hj535_2009]
- 紫外_可见分光光度法练习试题
- 止血海绵中微量甲醛酚试剂分光光度法测定
- 止血海绵中微量甲醛酚试剂分光光度法测定论文
- 用分光光度法测定复方药物安痛定片、阿司匹林和感克片中非那西丁含量论文
- 紫外分光光度法测定紫云英蜜中总酚的含量论文
- 分析化学课件紫外可见光分光光度法
- 分光光度法测定阿奇霉素片的含量论文
- 可见光及紫外光分光光度法测定丁香总黄酮含量的研究
- 紫外分光光度法测定峰胶胶囊中总黄酮的含量