

- 1.33 MB
- 9页

- 1、本文档共5页,可阅读全部内容。
- 2、本文档由网友投稿或网络整理,如有侵权请及时联系我们处理。
'第35卷第2期PreciousMetalsVol.35,No.2碱熔蒸馏分离-催化分光光度法测定超痕量锇、钌来新泽1,王琳1*,牛娜2,王敏捷1,来克冰1(1.河南省岩石矿物测试中心国土资源部贵金属分析与勘查技术重点实验室,郑州450012;2.辽宁省地质矿产研究院,沈阳110032)摘要:研制了新型的锇、钌混合氧化剂K2Cr2O7-NaBrO3和锇稀释剂As2O3-H2SO4,能够降低碱熔蒸馏分离-催化分光光度法测定锇、钌的全流程空白,提高了锇、钌催化As3+-Ce4+体系的灵敏度。在35℃时,通过延长反应时间,提高了方法的稳定性和降低方法的检测下线。使Os、Ru检出限(3σ)分别达到0.010ng/g和0.012ng/g。改进后的蒸馏装置使锇、钌蒸馏的安全性和工作效率极大提高。方法操作简便、效率高、成本低。测定国家铂族元素地球化学一级标准物质中的Os、Ru,相对误差(RE)为-18.0%~+4.40%,12次测定的相对标准偏差(RSD)均小于20.1%,满足地球化学调查样品分析质量要求。关键词:分析化学;蒸馏分离;催化分光光度法;超痕量;锇;钌中图分类号:O657.32文献标识码:A文章编号:1004-0676(2014)02-0050-09DeterminationofUltraTraceOsmiumandRutheniumbyAlkalineFusionDistillationSeparation-CatalyticSpectrophotometricMethodLAIXinze1,WANGLin1*,NIUNa2,WANGMinjie1,LAIKebing1(1.HenanRockMineralTestingCenters,KeyLabofMinistryofLandandResourcesAnalysisandExplorationofPreciousMetals,Zhenzhou450012,China;2.TheGeologyMineralInstituteforResearchofLiaoningProvince,Shenyang110032,China)Abstract:BydevelopinganewtypemixedoxidantofOsandRu,K2Cr2O7-NaBrO3anddiluentAs2O3-H2SO4ofOs,thefullprocedureblankinthedeterminationofRuandOsbyalkalifusiondistillationseparation-catalyticspectro-photometricmethodwasreduced,andthesensitivitiesofOs,RuinAs3+-Ce4+catalyticsystemwereimproved.Thestabilityofthemethodwasimprovedandthedetectionlimitwasreducedbyprolongingthereactiontimeat35℃.Detectedlimits(3σ)ofOsandRuwere0.010ng/gand0.012ng/g,respectively.Theresultsshowedthatthesafetyandefficiencyofdistillationweregreatlyimprovedbyimprovethedistillationdevice.Themethodissimpleoperation,highefficiencyandlowcost.ThemethodwasappliedtomeasureOsandRuinNationalPGEgeochemistrystandardsubstance.Therelativeerror(RE)was-18.0%~+4.40%,andtherelativestandarddeviation(RSD)of12-timesresultswaslessthan20.1%.Themethodcouldmeettheanalysisqualityrequirementsofgeochemicalsurveysample.Keywords:analyticalchemistry;distillationseparation;catalyticspectrophotometricmethod;ultratrace;osmium;ruthenium收稿日期:2013-07-15基金项目:国土资源部公益性行业科研专项经费项目(201211081-03-34)。第一作者:来新泽,男,工程师,研究方向:贵金属分析方法研究。E-mail:laixinze@126.com*通讯作者:王琳,女,高级工程师,研究方向:贵金属分析方法研究。E-mail:wanglin0630@126.com
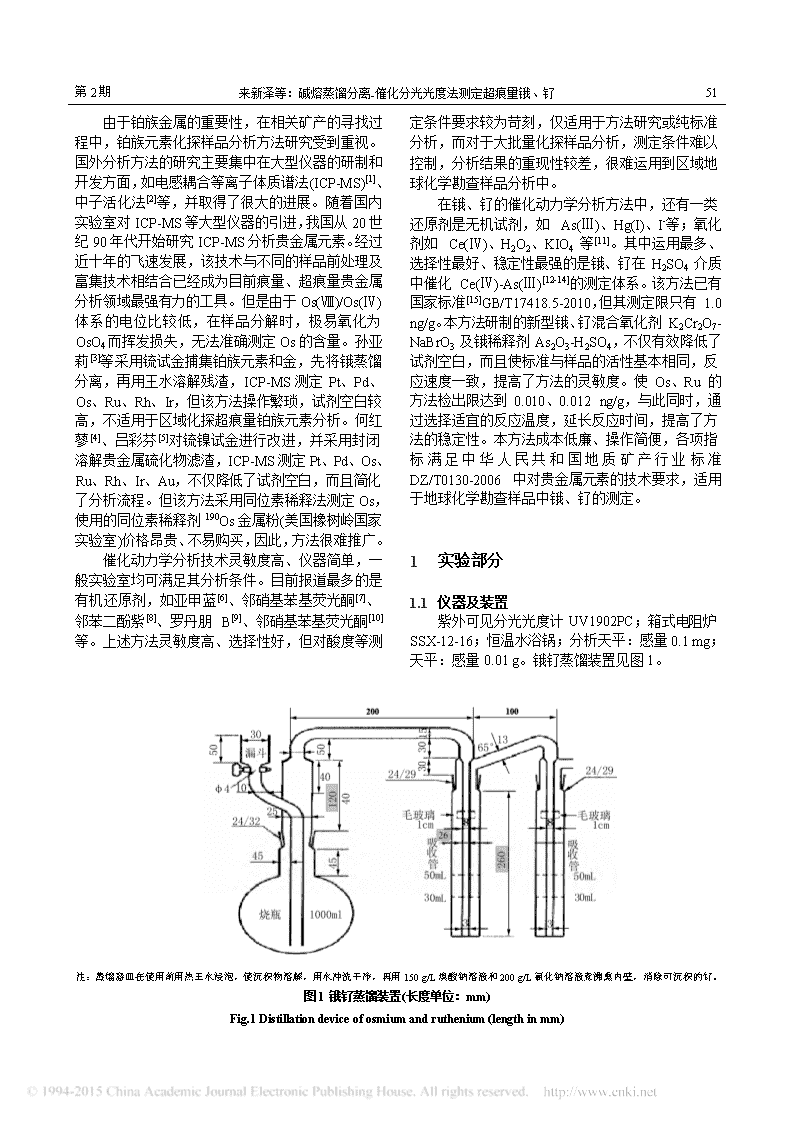
第2期51来新泽等:碱熔蒸馏分离-催化分光光度法测定超痕量锇、钌由于铂族金属的重要性,在相关矿产的寻找过程中,铂族元素化探样品分析方法研究受到重视。国外分析方法的研究主要集中在大型仪器的研制和开发方面,如电感耦合等离子体质谱法(ICP-MS)[1]、中子活化法[2]等,并取得了很大的进展。随着国内实验室对ICP-MS等大型仪器的引进,我国从20世纪90年代开始研究ICP-MS分析贵金属元素。经过近十年的飞速发展,该技术与不同的样品前处理及富集技术相结合已经成为目前痕量、超痕量贵金属分析领域最强有力的工具。但是由于Os(Ⅷ)/Os(Ⅳ)体系的电位比较低,在样品分解时,极易氧化为OsO4而挥发损失,无法准确测定Os的含量。孙亚莉[3]等采用锍试金捕集铂族元素和金,先将锇蒸馏分离,再用王水溶解残渣,ICP-MS测定Pt、Pd、Os、Ru、Rh、Ir,但该方法操作繁琐,试剂空白较高,不适用于区域化探超痕量铂族元素分析。何红蓼[4]、吕彩芬[5]对锍镍试金进行改进,并采用封闭溶解贵金属硫化物滤渣,ICP-MS测定Pt、Pd、Os、Ru、Rh、Ir、Au,不仅降低了试剂空白,而且简化了分析流程。但该方法采用同位素稀释法测定Os,使用的同位素稀释剂190Os金属粉(美国橡树岭国家实验室)价格昂贵、不易购买,因此,方法很难推广。催化动力学分析技术灵敏度高、仪器简单,一般实验室均可满足其分析条件。目前报道最多的是有机还原剂,如亚甲蓝[6]、邻硝基苯基荧光酮[7]、邻苯二酚紫[8]、罗丹朋B[9]、邻硝基苯基荧光酮[10]等。上述方法灵敏度高、选择性好,但对酸度等测定条件要求较为苛刻,仅适用于方法研究或纯标准分析,而对于大批量化探样品分析,测定条件难以控制,分析结果的重现性较差,很难运用到区域地球化学勘查样品分析中。在锇、钌的催化动力学分析方法中,还有一类还原剂是无机试剂,如As(Ⅲ)、Hg(I)、I-等;氧化剂如Ce(Ⅳ)、H2O2、KIO4等[11]。其中运用最多、选择性最好、稳定性最强的是锇、钌在H2SO4介质中催化Ce(Ⅳ)-As(Ⅲ)[12-14]的测定体系。该方法已有国家标准[15]GB/T17418.5-2010,但其测定限只有1.0ng/g。本方法研制的新型锇、钌混合氧化剂K2Cr2O7-NaBrO3及锇稀释剂As2O3-H2SO4,不仅有效降低了试剂空白,而且使标准与样品的活性基本相同,反应速度一致,提高了方法的灵敏度。使Os、Ru的方法检出限达到0.010、0.012ng/g,与此同时,通过选择适宜的反应温度,延长反应时间,提高了方法的稳定性。本方法成本低廉、操作简便,各项指标满足中华人民共和国地质矿产行业标准DZ/T0130-2006中对贵金属元素的技术要求,适用于地球化学勘查样品中锇、钌的测定。1实验部分1.1仪器及装置紫外可见分光光度计UV1902PC;箱式电阻炉SSX-12-16;恒温水浴锅;分析天平:感量0.1mg;天平:感量0.01g。锇钌蒸馏装置见图1。注:蒸馏器皿在使用前用热王水浸泡,使沉积物溶解,用水冲洗干净,再用150g/L溴酸钠溶液和200g/L氯化钠溶液煮沸熏内壁,消除可沉积的钌。图1锇钌蒸馏装置(长度单位:mm)Fig.1Distillationdeviceofosmiumandruthenium(lengthinmm)
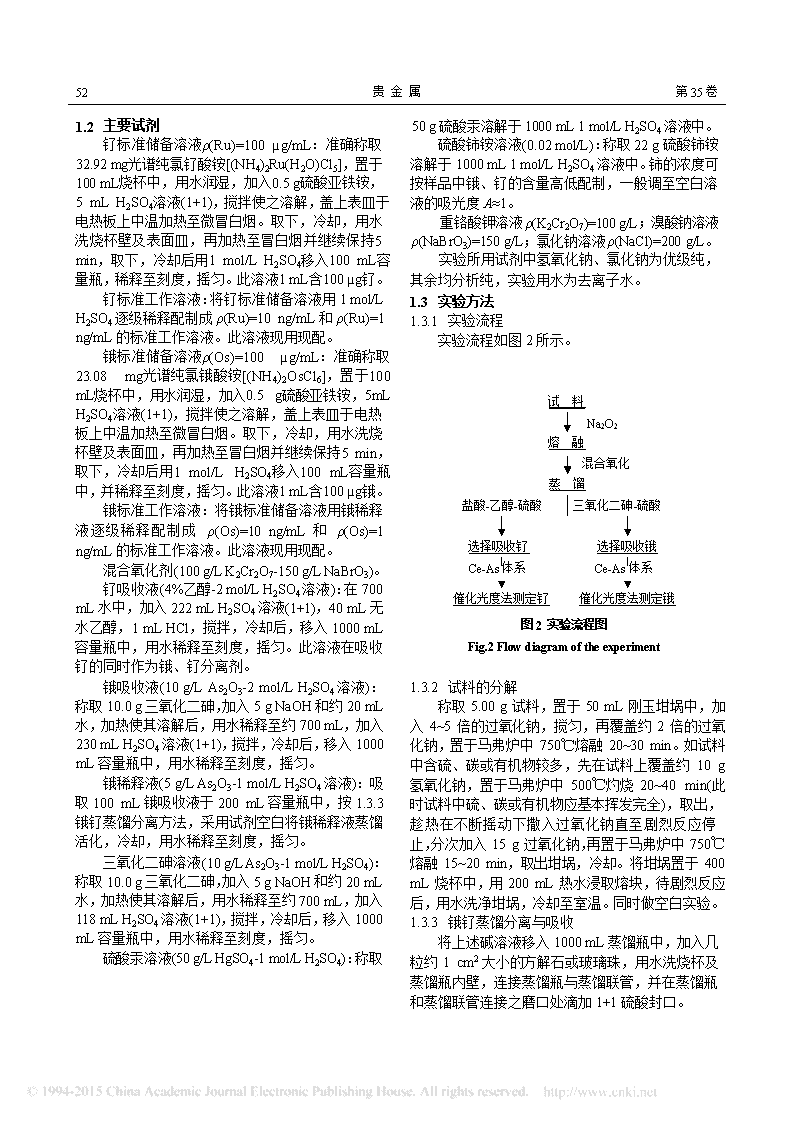
52贵金属第35卷1.2主要试剂钌标准储备溶液ρ(Ru)=100µg/mL:准确称取32.92mg光谱纯氯钌酸铵[(NH4)2Ru(H2O)Cl5],置于100mL烧杯中,用水润湿,加入0.5g硫酸亚铁铵,5mLH2SO4溶液(1+1),搅拌使之溶解,盖上表皿于电热板上中温加热至微冒白烟。取下,冷却,用水洗烧杯壁及表面皿,再加热至冒白烟并继续保持5min,取下,冷却后用1mol/LH2SO4移入100mL容量瓶,稀释至刻度,摇匀。此溶液1mL含100µg钌。钌标准工作溶液:将钌标准储备溶液用1mol/LH2SO4逐级稀释配制成ρ(Ru)=10ng/mL和ρ(Ru)=1ng/mL的标准工作溶液。此溶液现用现配。锇标准储备溶液ρ(Os)=100µg/mL:准确称取23.08mg光谱纯氯锇酸铵[(NH4)2OsCl6],置于100mL烧杯中,用水润湿,加入0.5g硫酸亚铁铵,5mLH2SO4溶液(1+1),搅拌使之溶解,盖上表皿于电热板上中温加热至微冒白烟。取下,冷却,用水洗烧杯壁及表面皿,再加热至冒白烟并继续保持5min,取下,冷却后用1mol/LH2SO4移入100mL容量瓶中,并稀释至刻度,摇匀。此溶液1mL含100µg锇。锇标准工作溶液:将锇标准储备溶液用锇稀释液逐级稀释配制成ρ(Os)=10ng/mL和ρ(Os)=1ng/mL的标准工作溶液。此溶液现用现配。混合氧化剂(100g/LK2Cr2O7-150g/LNaBrO3)。钌吸收液(4%乙醇-2mol/LH2SO4溶液):在700mL水中,加入222mLH2SO4溶液(1+1),40mL无水乙醇,1mLHCl,搅拌,冷却后,移入1000mL容量瓶中,用水稀释至刻度,摇匀。此溶液在吸收钌的同时作为锇、钌分离剂。锇吸收液(10g/LAs2O3-2mol/LH2SO4溶液):称取10.0g三氧化二砷,加入5gNaOH和约20mL水,加热使其溶解后,用水稀释至约700mL,加入230mLH2SO4溶液(1+1),搅拌,冷却后,移入1000mL容量瓶中,用水稀释至刻度,摇匀。锇稀释液(5g/LAs2O3-1mol/LH2SO4溶液):吸取100mL锇吸收液于200mL容量瓶中,按1.3.3锇钌蒸馏分离方法,采用试剂空白将锇稀释液蒸馏活化,冷却,用水稀释至刻度,摇匀。三氧化二砷溶液(10g/LAs2O3-1mol/LH2SO4):称取10.0g三氧化二砷,加入5gNaOH和约20mL水,加热使其溶解后,用水稀释至约700mL,加入118mLH2SO4溶液(1+1),搅拌,冷却后,移入1000mL容量瓶中,用水稀释至刻度,摇匀。硫酸汞溶液(50g/LHgSO4-1mol/LH2SO4):称取50g硫酸汞溶解于1000mL1mol/LH2SO4溶液中。硫酸铈铵溶液(0.02mol/L):称取22g硫酸铈铵溶解于1000mL1mol/LH2SO4溶液中。铈的浓度可按样品中锇、钌的含量高低配制,一般调至空白溶液的吸光度A≈1。重铬酸钾溶液ρ(K2Cr2O7)=100g/L;溴酸钠溶液ρ(NaBrO3)=150g/L;氯化钠溶液ρ(NaCl)=200g/L。实验所用试剂中氢氧化钠、氯化钠为优级纯,其余均分析纯,实验用水为去离子水。1.3实验方法1.3.1实验流程实验流程如图2所示。试料Na2O2熔融混合氧化馏蒸盐酸-乙醇-硫酸三氧化二砷-硫酸选择吸收钌选择吸收锇Ce-As体系Ce-As体系催化光度法测定钌催化光度法测定锇图2实验流程图Fig.2Flowdiagramoftheexperiment1.3.2试料的分解称取5.00g试料,置于50mL刚玉坩埚中,加入4~5倍的过氧化钠,搅匀,再覆盖约2倍的过氧化钠,置于马弗炉中750℃熔融20~30min。如试料中含硫、碳或有机物较多,先在试料上覆盖约10g氢氧化钠,置于马弗炉中500℃灼烧20~40min(此时试料中硫、碳或有机物应基本挥发完全),取出,趁热在不断摇动下撒入过氧化钠直至剧烈反应停止,分次加入15g过氧化钠,再置于马弗炉中750℃熔融15~20min,取出坩埚,冷却。将坩埚置于400mL烧杯中,用200mL热水浸取熔块,待剧烈反应后,用水洗净坩埚,冷却至室温。同时做空白实验。1.3.3锇钌蒸馏分离与吸收将上述碱溶液移入1000mL蒸馏瓶中,加入几粒约1cm2大小的方解石或玻璃珠,用水洗烧杯及蒸馏瓶内壁,连接蒸馏瓶与蒸馏联管,并在蒸馏瓶和蒸馏联管连接之磨口处滴加1+1硫酸封口。
第2期53来新泽等:碱熔蒸馏分离-催化分光光度法测定超痕量锇、钌在第一吸收管中准确加入25mL钌吸收液,第二吸收管中准确加入25mL锇吸收液,用蒸馏联管将第一、第二吸收管联结起来,接口处涂抹几滴1+1硫酸封口(见图1)。浓度以标准系列中零点的吸光度为1.000做依据来确定,如试料中锇、钌含量过高时,可加大硫酸铈铵的浓度。硫酸铈铵的用量一定要准确加入。1.3.5测定1.3.5.1钌的测定移取1~5mL第一吸收管中溶液,于25mL比色管中(不足5mL时,补加1mol/LH2SO4至5mL),加入2mL三氧化二砷溶液,1mL硫酸汞溶液,摇匀,将比色管连同比色管架浸入35℃恒温水浴中20min(夏季常温放置20min)。迅速加入1.00mL硫酸铈铵溶液,摇匀。同标准一起置于35℃恒温水浴中,以下与标准工作曲线测定步骤相同,以水作参比,在420nm波长处测定溶液的吸光度Ac,求lg(A0/Ac)值。从工作曲线上查出含钌量,计算结果。1.3.5.2锇的测定移取1~5mL第二吸收管中溶液,于25mL比色管中(不足5mL时,补加锇稀释液至5mL),加入2mL1mol/LH2SO4溶液,1mL硫酸汞溶液,摇匀,将比色管连同比色管架浸入35℃恒温水浴中20min(夏季常温放置20min)。迅速加入1.00mL硫酸铈铵溶液,摇匀。以下操作按1.3.5.1的测定步骤,得到锇含量结果。从蒸馏联管侧边漏斗加入120mLH2SO4溶液(1+1),16mL100g/L重铬酸钾溶液、4mL150g/L溴酸钠溶液和2滴200g/L氯化钠溶液,用水洗漏斗,关闭活塞。摇动蒸馏瓶使沉淀完全溶解。此时蒸馏瓶中应为黄色澄清透明溶液。将蒸馏瓶架于可调电炉上,第二吸收管浸入冷水槽中(冷水高度需达到吸收管50mL刻度线),加热蒸馏,待溶液沸腾后调节温度使溶液保持微沸,蒸馏至第二吸收管内溶液增至37~40mL时,取下蒸馏联管和吸收系统,将吸收管置于冷水中冷却至室温,用水冲洗蒸馏联管并稀释至50mL,摇匀。第一吸收管中溶液用于测定钌,第二吸收管中溶液用于测定锇。1.3.4工作曲线的配制1.3.4.1钌工作曲线的配制移取0.1ng/mL钌标准工作溶液0、0.2、0.5、1.0、1.5、2.0、2.5mL,分别置于一组25mL比色管中,补加1mol/LH2SO4至5mL,加入2mL三氧化二砷溶液,1mL硫酸汞溶液,摇匀,将比色管连同比色管架浸入35℃恒温水浴20min(夏季于常温放置20min),迅速加入1.00mL硫酸铈铵溶液,摇匀,继续置于35℃恒温水浴一定时间(以工作曲线中最高钌量的吸光值降至0.3附近时所需的时间来确定,一般在2.5~3h之间),取出,冷却,移入1cm比色皿中,以水作参比,在420nm波长处测定溶液的吸光度Ac,求lg(A0/Ac)值(工作曲线中零点的吸光度作为A0)。以钌的量作横坐标,lg(A0/Ac)值作纵坐标,绘制工作曲线。1.3.4.2锇工作曲线的配制移取0.1ng/mL锇标准工作溶液0、0.2、0.5、1.0、1.5、2.0、2.5mL,分别置于一组25mL比色管中,补加锇稀释液至5mL,加入2mL1mol/LH2SO4溶液,1mL硫酸汞溶液,摇匀,将比色管连同比色管架浸入35℃恒温水浴中20min(夏季常温放置20min),迅速加入1.00mL硫酸铈铵溶液,摇匀,继续置于35℃恒温水浴一定时间(以工作曲线中最高锇量的吸光值降至0.3附近时所需的时间来确定,一般在2.5h左右),取出,冷却,移入1cm比色皿中,以水作参比,以下测定同1.3.4.1。由于硫酸铈铵的浓度是可变的,一般来说,其2结果与讨论2.1坩埚的选择采用过氧化钠熔解贵金属时,通常使用的坩埚有铁坩埚、镍坩埚和刚玉坩埚。镍中常伴有铂族金属(尤其是钌),一般测定痕量、超痕量锇、钌时不选用。分别取上述坩埚,按实验方法加入同厂家、同批次、等量试剂平行测定20次,考察不同坩埚锇、钌空白值。从表1可知,铁坩埚对测定1ng/g以上的锇、钌影响不大,但对测定1ng/g以下的锇、钌,其空白值对测定结果影响较大,尤其对0.0xng/g的锇、钌,结果不准确。试验发现,刚玉坩埚的空白值远远低于铁坩埚和镍坩埚,因此,选择刚玉坩埚分解试料中锇、钌。表1不同坩埚空白值比较(n=20)Tab.1Comparisonofdifferentcrucibleblankvalues(n=20)元素空白值铁坩埚镍坩埚刚玉坩埚Ru/(ng/g)Os/(ng/g)0.0790.0420.4260.0560.0100.008注:使用相同量Na2O2、H2SO4、K2Cr2O7、NaBrO3和相应坩埚的平均空白值。
54贵金属第35卷2.2蒸馏装置的改进原蒸馏装置中,边管漏斗与吸收支管间有一磨口,在蒸馏时,RuO4易沉积于此造成损失。而且在蒸馏初期,烧瓶内的过氧化钠碱溶液遇硫酸溶液放出大量的过氧化氢,此气体遇热迅速膨胀,烧瓶内压力过大,致使接口冲开,烧瓶内溶液喷射,锇、钌挥发。本方法将此处连接口连为一体,并将其由原80mm加长至120mm(图1中灰色部分),既增大了蒸馏的安全系数,又防止吸附锇、钌的损失。另外,为了防止第一吸收管中的溶液冲至第二吸收管中,把吸收管的长度由250mm加长至260mm,并将吸收管直径由28mm减少至26mm(图1中灰色部分),并且控制两吸收管间支管的角度在65°~70°范围内。两吸收管间距不小于100mm。2.3氧化剂选择、配比及用量Os8+/Os4+的氧化还原电位为1.00V,Ru8+/Ru4+的氧化还原电位为1.4V。一般地说,凡是氧化还原电位高于1.4V的氧化剂均有可能将Ru、Os从溶液中同时蒸馏出来。氧化还原电位高于1.0V低于1.4V的氧化剂,能选择性地蒸馏锇。蒸馏锇、钌的有效氧化剂有文献报道的见表2[11]。表3不同氧化剂的空白值(n=20)Tab.3Theblankvaluesofdifferentoxidants(n=20)元素空白值KMnO4NaBrO3KBrO3有大量Br有大量BrRu/(ng/g)Os/(ng/g)0.0460.0520.0060.003元素空白值NaBiO3K2Cr2O7KIO4Ru/(ng/g)Os/(ng/g)>0.x>0.x0.0050.0040.0050.006通过实验,研制出一种新型的锇钌混合氧化剂K2Cr2O7-NaBrO3。合适配比的混合氧化剂既能提高锇、钌的回收率,又不会析出干扰测定的物质,同时满足超痕量锇钌的分析要求。分别取10ng的Ru、Os标准溶液置于1000mL蒸馏瓶中,按表4加入不量的氧化剂进行蒸馏。从表4可知,随着混合氧化剂加入量的减少,回收率会有所降低,尤其是Os。但加入量太大时,其空白将会增大,甚至会有Br析出。本方法选择16mL100g/LK2Cr2O7和4mL150g/LNaBrO3。表4蒸馏Ru、Os混合氧化剂配比试验Tab.4MixedoxidantsfordistillationRu,OsK2Cr2O7NaBrO3RuOs表2蒸馏锇、钌的有效氧化剂Tab.2OxidantsfordistillationofOs,Ru序号用量/mL用量/mL回收率/%空白值/ng回收率/%空白值/ng123456789102525252020201515151054354354351041061031031051011021041011020.0260.0220.0230.0130.0150.0140.0100.0090.0080.006少量Br104102少量Br10310210110299.264.80.0320.0170.0150.0360.0100.0090.0180.0060.0050.010其中最常用的、氧化能力较强的,并且已应用在国标方法中的锇钌氧化剂为KMnO4。但对于超痕量锇钌分析,KMnO4的空白值已经超出地球化学勘查样品中锇、钌测定的要求。对几种主要的氧化剂平行测定20次,考察其空白值,结果列于表3。由表3可知,KBrO3易析出Br,干扰测定;KMnO4及NaBiO3的空白值较高,不能满足超痕量锇、钌分析需要;K2Cr2O7、NaBrO3、KIO4的空白值都比较低,但是,仅用K2Cr2O7或KIO4作氧化剂时,钌的回收率只有70%,锇的回收率还不到70%,仅用NaBrO3作氧化剂时,也会分解出大量的Br,干扰测定。111041030.00562.10.004在蒸馏时,还需加入数滴氯化钠(浓度200g/L),否则混合氧化剂的氧化能力会降低;但也不宜加入太多,因为当气温高时,过多氯化钠易造成锇不稳定。2.4As-Ce体系最佳波长的选择波长选择实验结果如图3~5所示。从图3锇、钌吸收曲线可知,As-Ce体系最大吸收峰在380nm处。但对一般的分光光度计而言,波长小于400nm时,吸光值不稳定,误差太大。蒸馏锇+钌(或钌)的氧化剂蒸馏锇的氧化剂HClO4+NaBiO3KMnO4+NaClK2Cr2O7+H2SO4KIO4+NaBrO3+NaClHClO4PbO2+NaBrO3+NaClNaBrO3K2Cr2O7+缩水磷酸Cl2+NaOHNaBiO3+KMnO4+NaBrO3+NaClH2O2+Ag2O3Ce4++缩水磷酸H2O2+HCl
第2期55来新泽等:碱熔蒸馏分离-催化分光光度法测定超痕量锇、钌2.5酸度对锇、钌催化As3+-Ce4+体系反应速度的影响分别选用0.5、1.0、1.5、2.0mol/L硫酸介质,考察其对锇、钌反应速度的影响。结果如图所示。6、7图3锇、钌的吸收曲线Fig.3AbsorptioncurveofOs,Ru从图4、5不同波长锇、钌的线性曲线可知,波长越短,灵敏度越高,但线形较差,尤其是锇的表现更为突出。因此,选择420nm为最佳吸收波长。图6酸度对Ru反应速度的影响Fig.6EffectofacidityonRureactionrate(1.0.5mol/LH2SO4;2.1mol/LH2SO4;3.1.5mol/LH2SO4;4.2mol/LH2SO4)图4钌最佳波长选择Fig.4OptimalwavelengthselectionofRu(1.420nm0.5mol/LH2SO4;2.400nm1mol/LH2SO4;3.410nm1mol/LH2SO4;4.420nm1mol/LH2SO4)图7酸度对Os反应速度的影响Fig.7EffectofacidityonOsreactionrate(1.0.5mol/LH2SO4;2.1mol/LH2SO4;3.1.5mol/LH2SO4;4.2mol/LH2SO4)由图6、7可知,体系酸度越小,反应速度越快,灵敏度越高。但当体系酸度低至0.5mol/L时,虽然反应速度大大提高,但曲线线性关系较差。因此,选择1mol/L的硫酸酸度。2.6As、Ce用量对锇钌催化As3+-Ce4+反应速度的影响在相同的酸度、温度、时间条件下,固定Ce4+的加入量(0.02mol/L硫酸铈铵1.00mL),改变As3+的浓度,即选择不同的[As3+]/[Ce4+]比,考察As、Ce用量对锇、钌催化As3+-Ce4+反应速度的影响,图5锇最佳波长选择Fig.5OptimalwavelengthselectionofOs(1.400nm1mol/LH2SO4;2.410nm1mol/LH2SO4;3.420nm0.5mol/LH2SO4;4.420nm1mol/LH2SO4)
56贵金属第35卷结果如图8、9所示。峭,线性关系被破坏,浓度范围也相应缩小。但温度太低,反应速度缓慢,曲线斜率太小。通过实验确定锇钌催化As3+-Ce4+反应的最佳温度为35℃,同时,根据锇钌的浓度选择催化反应时间,提高了方法的稳定性,并且降低了方法的检测下限。2.8方法的回收率试验于5.0g国家铂族元素地球化学一级标准物质GBW07289、GBW07294、GBW07340中,分别加入一定量的Os、Ru标准溶液,对其进行全流程回收率试验。由表5结果可知,Ru、Os回收率分别为94%~106%、90%~108%。图8[As3+]/[Ce4+]对Ru催化反应速度的影响Fig.8EffectofAs3+/Ce4+ratioonRucatalysisreactionrate(1.0.1mol/LAs2O3;2.0.075mol/LAs2O3;3.0.05mol/LAs2O3;4.0.04mol/LAs2O3;5.0.025mol/LAs2O3)表5样品加标准回收率Tab.5Therecoverytestsofthemethod标准值加入量测得量回收量回收率元素标样号/(ng/g)/(ng/g)/(ng/g)/(ng/g)/%GBW07289GBW07294GBW073400.100.660.430.100.500.500.1971.130.960.0970.470.539794.0106RuGBW07289GBW07294GBW073400.060.640.250.100.500.500.1621.090.790.1020.450.5410290108Os2.9方法检出限、精密度和准确度锇、钌在一定的价态、介质中才具备活性,且其活性的大小也是随条件的变化而变化。对于痕量锇、钌分析,只要锇、钌标准的介质与蒸馏样品的介质相同,且活性基本一致,对测定结果基本无影响。但对于超痕量锇、钌分析而言,锇、钌标准与蒸馏样品的活性稍有差别,其反应速度就不同步,结果差别很大。取0.00、0.02、0.05、0.10、0.15、0.20、0.25ng两组相同的Os标准溶液,其中一组采用未经活化的锇稀释剂稀释至5mL,另一组采用经活化的锇稀释剂(本实验研究)稀释至5mL进行相同的实验。经过活化的锇稀释剂的一组Os标准溶液,水浴2.5h后吸光值满足要求,并且标准曲线线性良好,与样品反应速度基本一致;而另一组Os标准溶液,水浴4h后标准曲线零点的吸光值才近似为1,曲线斜率较小。这证明使用活化后的锇稀释剂,标准系列与蒸馏样品活性相近、反应速度一致,灵敏度较高,不仅提高了结果的准确度和精密度,而且有效降低了全流程空白,使Os的检出限达到0.010ng/g,Ru的检出限达到0.012ng/g。称取一定量国家铂族元素地球化学一级标准物图9[As3+]/[Ce4+]对Os催化反应速度的影响Fig.9EffectofAs3+/Ce4+ratioonOscatalysisreactionrate(1.0.1mol/LAs2O3;2.0.075mol/LAs2O3;3.0.05mol/LAs2O3;4.0.04mol/LAs2O3;5.0.025mol/LAs2O3)由图8、9可知,As3+-Ce4+反应速度随[As3+]浓度的增大而加快,即As3+-Ce4+反应速度随[As3+]/[Ce4+]比值增加而增加。但当As2O3浓度增大到0.075mol/L时,曲线开始弯曲,偏向横轴,Ru更为明显,且As2O3浓度越大,弯曲的程度越大。当As2O3的浓度为0.04~0.05mol/L时,其线性关系良好,灵敏度较高。选择As用量为0.05mol/L的As2O32mL,Ce用量为0.02mol/L的硫酸铈铵1.00mL,总体积为9mL,测定范围为0~0.5ng。2.7温度、时间对反应速度的影响一般来说,温度高则催化时间短,温度低则催化时间长。如果温度过高,反应速度过快,曲线陡
第2期57来新泽等:碱熔蒸馏分离-催化分光光度法测定超痕量锇、钌质GBW07288、GBW07289、GBW07294、GBW07291,按照样品分析步骤,平行测定12次,精密度、准确度结果见表6。表6方法的精密度和准确度(n=12)Tab.6Precisionandaccuracytestsofthemethod(n=12)分析标准值平均值相对误差RE/%相对标准偏差RSD/%标样号元素/ng/ngRuOsRuOsRuOsRuOs0.050.050.100.060.660.642.52.40.0520.0410.0940.0620.680.652.612.32+4.00-18.0-6.00+3.33+3.03+1.56+4.40-3.3318.920.115.817.412.78.976.285.23GBW07288图11中国南方锇地球化学图Fig.11GeochemicalmapsofOsinsouthernChinaGBW07289GBW07294目前这一方法正用于承担中国地质科学院地球物理地球化学勘查研究所《全国地球化学基准值建立》项目中3万余件土壤、岩石样品中Os、Ru的分析测试,方法检出限、精密度、准确度及报出率均满足要求,其分析数据为《全国地球化学基准值建立》提供理论依据。GBW07291表6结果表明,Ru的精密度(RSD)为6.28%~18.9%、Os的精密度(RSD)为5.23%~20.1%,Ru的准确度(RE)为-6.00%~+4.40%、Os的准确度(RE)为-18.0%~+3.33%,满足区域地球化学调查样品的分析要求。2.10方法的应用3结语采用碱熔-重铬酸钾-溴酸钠做混合氧化剂,As3+-Ce4+体系催化光度法测定Os、Ru,有效控制全测试过程中的空白值,该分析方法具有灵敏度高、检出限低、效率高和成本低的特点。采用本方法已完成地球化学勘查样品4万余件,其中完成谢学锦院士全国低密度地球化学样品6000余件,成功编制了首幅中国南方(云南、贵州、四川、广西等12个省)锇、钌地球化学图(图10、11)。参考文献:[1]DateAR,DavisAE,CheungYY.Thepotentialoffireassayandinductivelycoupledplasmasourcemassspectrometryforthedeterminationofplatinumgroupelementsingeologicalmaterials[J].Analyst,1987,112(9):1217-1222.ParrySJ,AsifM,SinclairIW.Radiochemicalfire-assayfordeterminationoftheplatinum-groupelements[J].JRadioanalNuclChem,1988,123(2):593-606.孙亚莉,管希云,杜安道.锍试金富集贵金属元素I.等离子体质谱法测定地质样品中痕量铂族元素[J].岩矿测试,1997,16(1):12-17.[2][3]图10中国南方钌地球化学图Fig.10GeochemicalmapsofRuinsouthernChina
58贵金属第35卷SunY,GuanX,DuA.PreconcentrationofpreciousmetalelementsbynickelsulphidefireassayI.DeterminationofplatinumgroupelementsingeologicalsamplesbyICP-MS[J].RockandMineralAnalysis,1997,16(1):12-17.何红蓼,吕彩芬,周肇茹,等.锍镍试金-等离子体质谱法测定地球化学勘查样品中的铂族元素和金I.分析流程的简化[J].岩矿测试,2001,20(3):191-194.HeH,LvC,ZhouZ,etal.Determinationofplatinumgroupelementsandgoldingeochemicalexplorationsamplesbynickelsulphidefireassay-ICP-MSI.Simplificationoftheanalyticalprocedure[J].RockandMineralAnalysis,2001,20(3):191-194.吕彩芬,何红蓼,周肇茹,等.锍镍试金-等离子体质谱法测定地球化学勘查样品中的铂族元素和金Ⅱ.分析流程空白的降低[J].岩矿测试,2002,21(1):7-11.LuC,HeH,ZhouZ,etal.Determinationofplatinumgroupelementsandgoldingeochemicalexplorationsamplesbynickelsulfidefireassay-ICP-MSII.Reductionofreagentblank[J].RockandMineralAnalysis,2002,21(1):7-11.李祖碧,曹秋娥,王加林,等.高碘酸盐氧化亚甲蓝动力学光度法测定痕量锇[J].分析化学,2000,28(3):361-364.LiZ,CaoQ,WangJ,etal.Kineticspectrophotometricdeterminationoftraceosmiumbyitscatalyticeffectontheoxidationofmethlyeneblue[J].ChineseJournalofAnalyticalChemistry,2000,28(3):361-364.张庆合,赵中一,金继红,等.催化褪色光度法测定痕量锇—邻硝基苯基荧光酮-锇-H2O2体系[J].理化检验:化学分册,1994,30(6):335-336.ZhangQ,ZhaoZ,JingJ,etal.Photometricdeterminationoftraceamountsofosmiumthroughitscatalyticeffectontheoxidationofo-NO2-PFbyH2O2[J].PhysicalTestingandChemicalAnalysisPartB:ChemicalAnalysis,1994,30(6):335-336.唐宁莉,唐建洪.催化光度法测定痕量锇[J].岩矿测试,1996,15(4):283-285.TangN,TangJ.Catalyticspectrophotometricdeter-minationoftraceosmium[J].RockandMineralAnalysis,1996,15(4):283-285.蒋治良.超痕量钌的催化光度分析—KIO4-罗丹明B新体系[J].化学通报,1992(1):38-40.周之荣,张丽珍,王群,等.过氧化氢氧化邻硝基苯基荧光酮褪色反应动力学光度法测定痕量钌[J].冶金分析,2008,28(12):66-69.ZhouZ,ZhangL,WangQ,etal.Kineticspectro-photometricdeterminationoftracerutheniumbasedonoxidationofo-nitrophenylfluoronewithhydrogenperoxide[J].MetallurgicalAnalysis,2008,28(12):66-69.董守安.现代贵金属分析[M].北京:化学工业出版社,2007.郭漾光.催化比色测定矿石中的锇和钌[J].贵金属,1981,2(2):22-28.王琳.炭质页岩及矿化炭质页岩中锇钌的测定[J].黄金,2004,25(6):50-52.WangL.Determinationofosmiumandrutheniumincarbonaceousshaleandmineralizedcarbonaceousshale[J].Gold,2004,25(6):50-52.王琳,来新泽,吴建政,等.地球化学勘探样品中超痕量锇、钌分析方法研究[J].贵金属,2004,25(3):49-53.WangL,LaiX,WuJ,etal.Determinationofultra-tracerutheniumandosmiumingeochemicalexplorationsamples[J].PreciousMetals,2004,25(3):49-53.郑存江,胡勇平,刘清辉.GB/T17418.5-2010地球化学样品中贵金属分析方法第5部分:钌量和锇量蒸馏分离-催化分光光度法[S].北京:中国标准出版社,2011.[8][4][9][10][5][11][12][13][6][14][7][15]'
您可能关注的文档
- 紫外分光光度法测氧氟沙星片剂中氧氟沙星的含量
- 邻二氮杂菲分光光度法测定铁_实验报告(精)
- 紫外分光光度法在药物分析中的应用
- 4-氨基安替比林萃取分光光度法测定水中挥发酚的质量控制指标研究
- 火焰原子吸收分光光度法测定爆米花中铅含量
- DB12T 847-2018 饲料中总糖的测定 分光光度法
- DB13T 1090-2009 饲料用酶制剂中木聚糖酶活力的测定 分光光度法
- DB13T 1091-2009 饲料用酶制剂中酸性蛋白酶活力的测定 分光光度法
- DB13T 1095-2009 饲料用酶制剂中α-淀粉酶活力的测定 分光光度法
- DB51T 1347-2011 作物中镉的测定 石墨炉原子吸收分光光度法
- GB-T 16129-1995 居住区大气中甲醛卫生检验标准方法 分光光度法
- GBT 11912-1989 水质 镍的测定 火焰原子吸收分光光度法
- GBT 13196-1991 水质 硫酸盐的测定 火焰原子吸收分光光度法
- GBT 13903-1992 水质 梯恩梯的测定 分光光度法
- GBT 13900-1992 水质 黑索今的测定 分光光度法
- GBT 13897-1992 水质 硫氰酸盐的测定 异烟酸一吡唑啉酮分光光度法
- GBT 14375-1993 水质 一甲基肼的测定 对二甲氨基苯甲醛分光光度法
- GBT 14376-1993 水质 偏二甲基肼的测定 氨基亚铁氰化钠分光光度法